Analysis Functions¶
This module defines MSA analysis functions.
- calcShannonEntropy(msa, ambiguity=True, omitgaps=True, **kwargs)[source]¶
Return Shannon entropy array calculated for msa, which may be an MSA instance or a 2D Numpy character array. Implementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types.
All non-alphabet characters are considered as gaps, and they are handled in two ways:
- non-existent, the probability of observing amino acids in a given column is adjusted, by default
- as a distinct character with its own probability, when omitgaps is False
- buildMutinfoMatrix(msa, ambiguity=True, turbo=True, **kwargs)[source]¶
Return mutual information matrix calculated for msa, which may be an MSA instance or a 2D Numpy character array. Implementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
Mutual information matrix can be normalized or corrected using applyMINormalization() and applyMICorrection() methods, respectively. Normalization by joint entropy can performed using this function with norm option set True.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
- calcMSAOccupancy(msa, occ='res', count=False)[source]¶
Return occupancy array calculated for residue positions (default, 'res' or 'col' for occ) or sequences ('seq' or 'row' for occ) of msa, which may be an MSA instance or a 2D NumPy character array. By default, occupancy [0-1] will be calculated. If count is True, count of non-gap characters will be returned. Implementation is case insensitive.
- applyMutinfoCorr(mutinfo, corr='prod')[source]¶
Return a copy of mutinfo array after average product correction (default) or average sum correction is applied. See [DSD08] for details.
[DSD08] Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 2008 24(3):333-340.
- applyMutinfoNorm(mutinfo, entropy, norm='sument')[source]¶
Apply one of the normalizations discussed in [MLC05] to mutinfo matrix. norm can be one of the following:
'sument':
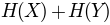 , sum of entropy of columns
, sum of entropy of columns'minent':
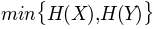 , minimum entropy
, minimum entropy'maxent':
 , maximum entropy
, maximum entropy- 'mincon':
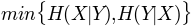 , minimum conditional
, minimum conditional entropy
- 'mincon':
- 'maxcon':
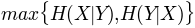 , maximum conditional
, maximum conditional entropy
- 'maxcon':
where
 is the entropy of a column, and
is the entropy of a column, and
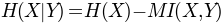 . Normalization with joint entropy, i.e.
. Normalization with joint entropy, i.e.
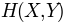 , can be done using buildMutinfoMatrix() norm
argument.
, can be done using buildMutinfoMatrix() norm
argument.[MLC05] Martin LC, Gloor GB, Dunn SD, Wahl LM. Using information theory to search for co-evolving residues in proteins. Bioinformatics 2005 21(22):4116-4124.
- calcRankorder(matrix, zscore=False, **kwargs)[source]¶
Returns indices of elements and corresponding values sorted in descending order, if descend is True (default). Can apply a zscore normalization; by default along axis - 0 such that each column has mean=0 and std=1. If zcore analysis is used, return value contains the zscores. If matrix is smymetric only lower triangle indices will be returned, with diagonal elements if diag is True (default).
- buildSeqidMatrix(msa, turbo=True)[source]¶
Return sequence identity matrix for msa.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
- uniqueSequences(msa, seqid=0.98, turbo=True)[source]¶
Return a boolean array marking unique sequences in msa. A sequence sharing sequence identity of sqid or more with another sequence coming before itself in msa will have a False value in the array.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
- buildOMESMatrix(msa, ambiguity=True, turbo=True, **kwargs)[source]¶
Return OMES (Observed Minus Expected Squared) covariance matrix calculated for msa, which may be an MSA instance or a 2D NumPy character array. OMES is defined as:
(N_OBS - N_EX)^2 (f_i,j - f_i * f_j)^2 OMES_(i,j) = sum(------------------) = N * sum(-----------------------) N_EX f_i * f_jImplementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
- buildSCAMatrix(msa, turbo=True, **kwargs)[source]¶
Return SCA matrix calculated for msa, which may be an MSA instance or a 2D Numpy character array.
Implementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
- buildDirectInfoMatrix(msa, seqid=0.8, pseudo_weight=0.5, refine=False, **kwargs)[source]¶
Return direct information matrix calculated for msa, which may be an MSA instance or a 2D Numpy character array.
Sequences sharing sequence identity of seqid or more with another sequence are regarded as similar sequences for calculating their weights using calcMeff().
pseudo_weight are the weight for pseudo count probability.
Sequences are not refined by default. When refine is set True, the MSA will be refined by the first sequence and the shape of direct information matrix will be smaller.
- calcMeff(msa, seqid=0.8, refine=False, weight=False, **kwargs)[source]¶
Return the Meff for msa, which may be an MSA instance or a 2D Numpy character array.
Since similar sequences in an msa decreases the diversity of msa, Meff gives a weight for sequences in the msa.
For example: One sequence in MSA has 5 other similar sequences in this MSA(itself included). The weight of this sequence is defined as 1/5=0.2. Meff is the sum of all sequence weights. In another word, Meff can be understood as the effective number of independent sequences.
Sequences sharing sequence identity of seqid or more with another sequence are regarded as similar sequences to calculate Meff.
Sequences are not refined by default. When refine is set True, the MSA will be refined by the first sequence.
The weight for each sequence are returned when weight is True.